Mutation free energy calculations
A. Introduction
Now we are entering the realm of protein design. We will take a small protein
and try to make it better. Or at least more stable. In order to achieve that,
we are going to carry out "alchemical" mutations during the simulations and
compute the corresponding free energy changes. If we do this both in the
folded and unfolded states, and compute the difference between them, this
will yield us an estimate of the difference in stability between wild type
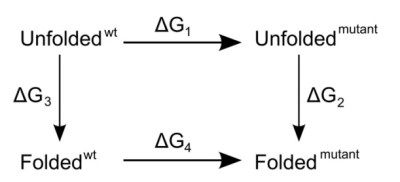
and mutant, as indicated in this thermodynamic cycle:
Experimentally, to address the same question, we would access the vertical
legs of the cycle and carry out folding/unfolding experiments to determine
ΔG3 and ΔG2. The difference ΔΔG =
ΔG3 - ΔG2 is then the difference in folding free
energy between wild type and mutant. In the simulations, this would be too
demanding, as even the folding time of a small peptide can be computationally
prohibitive, therefore preventing us from reliably obtaining the folding free
energies ΔG3 and ΔG2 directly from the
simulations for most peptides and proteins. However, we can relatively
accurately obtain the mutation free energies ΔG1 and
ΔG4 from "alchemical" mutations in which we morph one amino
acid into another and compute the according free energy change. If we do that
both in the folded and the unfolded states, we can make use of the fact that
the free energy is a state variable and therefore that the sum of all four
free energy values must be zero. Or, to put it differently, ΔΔG =
ΔG3 - ΔG2 = ΔG1 -
ΔG4. For the unfolded state, we do not really know what it
looks like. So we are going to assume that it can be modeled by a small piece
of unstructured peptide.
In the current tutorial we will focus on the simulation preparation of a folded protein,
however, an identical procedure can be applied to the short unstructed peptides as well.
In fact, the free energy differences for mutations
in GXG tripeptides can be precomputed for a given mutation library in a
specific force field.
Here you can find the precomputed ΔG values for GXG
tripeptides in several force fields.
In the next section, we will prepare a simulation for free energy calculations.
Further, we will demonstrate how to set-up the equilibrium slow-growth TI simulations
and the non-equilibrium free energy calculations.
For a more detailed description of the non-equilibrium simulation protocol have a look into
this book chapter.
In the last section of this tutorial we will analyze the obtained results.
Go back to Contents
B. System setup
 We are now going to set up the system with an alchemical mutation for the subsequent simulations.
The set up is a bit different from a usual gromacs set up, as we use the
pmx package for
this, which is an add-on to gromacs. We prepared an example based on a small
engineered mini-protein Trp-cage.
Of course, feel free to use any other protein of interest,
however, the further tutorial will concentrate on Trp-cage.
The next example assumes that you work with the
prepared "peptide.pdb". If not, please rename the filenames accordingly.
We are now going to set up the system with an alchemical mutation for the subsequent simulations.
The set up is a bit different from a usual gromacs set up, as we use the
pmx package for
this, which is an add-on to gromacs. We prepared an example based on a small
engineered mini-protein Trp-cage.
Of course, feel free to use any other protein of interest,
however, the further tutorial will concentrate on Trp-cage.
The next example assumes that you work with the
prepared "peptide.pdb". If not, please rename the filenames accordingly.
First, let us have a close look at the structure and see which mutation you'd
consider to be potentially stabilizing:
pymol peptide.pdb
You will see that the peptide is folded around a central tryptophan
residue. It might be tempting to mutate this residue, but remember that a
larger perturbation will take longer to converge in the simulation, so a
smaller or more conservative change might be more promising. In addition, it
is recommended not to introduce mutations that introduce charge
changes. This is due to the fact that the buildup of a net charge during the
morphing simulations can lead to artifacts and longer convergence times.
To define where we want the mutation and to set up the morph
structure we will use pmx mutate.py function.
Prior to doing that, several environmental variables need to be set:
GMXLIB variable should point to the pmx_source/data/mutff45 directory;
PYTHONPATH and LD_LIBRARY_PATH need to include the pmx installation path;
finally the pmx_source/scripts folder should be added to your PATH for convenience.
These environmental variables should already be properly set on the machines you are using.
To make sure of that you may type
echo $GMXLIB
This command will tell you where Gromacs and pmx will be searching for the force field files.
If you are using the prepared "peptide.pdb",
you are ready to run the mutation script.
However, in case you chose your own protein, it is important to prepare a clean version
of the structure file compatible with the Gromacs atom and residue naming.
For that you can run
gmx pdb2gmx -f peptide.pdb -o peptide.pdb -ff amber99sbmut -water tip3p -ignh
The command will tell you about any missing atoms or residues that need to be corrected.
Once all is set, run the mutate.py script:
mutate.py -f peptide.pdb -o mutant.pdb -ff amber99sbmut
Choose the residue you want to mutate and what you want to mutate it into.
Note that, as is frequently done in experiments, an alanine scan might be a
good starting point to identify promising locations.
For the further steps in the tutorial we will use S13A (serine to alanine) mutation.
If your are thinking of a different mutation, you may consider selecting a mutation for
which a tripeptide value has already been precomputed.
The terminal residues should not be selected at this step: to mutate terminal residues,
it would be necessary to add caps to the peptide.
Before going to the next step, investigate what changes have been introduced in the mutant.pdb file.
Once a mutation has been chosen, create a corresponding topology:
gmx pdb2gmx -f mutant.pdb -o conf.pdb -ff amber99sbmut -water tip3p
Here, the force-field used needs to be consistent with the force-field choice
from the previous step (amber99sbmut in this case).
We now have to post-process the topoloogy to add the morphing parameters:
generate_hybrid_topology.py -p topol.top -o newtop.top -ff amber99sbmut
In fact, these three steps of amino acid mutation and hybrid topology generation
could also have be carried out via a web-based interface.
Now we are ready to set up the simulation. First, we need to define a
simulation box:
gmx editconf -f conf.pdb -o box.pdb -bt dodecahedron -d 0.9
and we fill it with water:
gmx solvate -cp box -cs spc216 -o water.pdb -p newtop
To prepare the first energy minimization download the em.mdp file and run:
gmx grompp -f em.mdp -c water.pdb -p newtop.top
If you looked carefully, you'll see that grompp warned that the system carries
a net charge. In order to avoid artifacts due to a periodic charged system, we
add a chloride atom:
gmx genion -s -nn 1 -o ions.pdb -nname CL -pname NA -p
newtop.top
and select "13" as solvent group, so a water molecule can be replaced with a chloride ion. Now we can prepare for the energy minimization again:
gmx grompp -f em.mdp -c ions.pdb -p newtop.top -maxwarn 1
and carry out the actual energy minimization:
gmx mdrun -v -c em.pdb
Before we can set up the free energy calculation, we'll equilibrate the system
by running a short simulation of the wild type peptide (eq.mdp
file for equilibration):
echo 0 | gmx trjconv -s topol.tpr -f em.pdb -o em.pdb -ur compact -pbc mol
gmx grompp -f eq.mdp -c em.pdb -p newtop.top
gmx mdrun -v -ntomp 2 -nice 0 -c eq.gro -ntmpi 1
Depending on the way how Gromacs was compiled, you may not need the flag "-ntmpi 1".
Note that you can also use more (virtual) cores for the simulation by
providing a larger number after -ntomp.
Go back to Contents
C1. Slow-growth TI: simulation
Now we are ready to do the actual free energy calculation.
Firstly, we will use a slow-growth TI approach, where a transition from the WT to mutant
state will be performed by keeping the system in a quasi-equilibrium state.
Please have a look at the MD parameter file md.mdp. You will find a section
called "Free energy control" where you will see that free energy is switched
on, the initial lambda is chosen as zero, and that delta-lambda (per MD step)
is set such that at the end of the simulation lamda is at 1. Therefore, we are
using the slow-growth method here. Now start the actual simulation.
gmx grompp -f md.mdp -c eq.gro -p newtop.top
gmx mdrun -v -ntomp 2 -nice 0 -c md.gro -ntmpi 1
The simulation will take a while. Nevertheless, the number of steps defined in
"md.mdp" corresponds to only 100 ps, which is too short to expect a converged
result, and therefore for an accurate free energy estimate. Therefore, to
assess convergence, we suggest to carry out two validation steps:
- make the simulation two, five or ten times longer (or shorter) and check
if you arrive at the same result.
- do the backward transition and check if you get the same result (but then negative).
Go back to Contents
C2. Slow-growth TI: analysis
As already mentioned, slow-growth thermodynamic integration (TI) approach relies on an assumption that during a transition
the system remains in a quasi-equilibrium state.
Therefore, integration over the dH/dl curve directly yields a difference in free energy.
You may want to firstly analyze the results that were generated in the 100 ps simulation
that you just performed.
For a more converged estimate, we have also prepared
10 ns forward and backward transitions of an S13A mutation
in the TRP cage protein.
Integration can be done using the gmx analyze tool. (Keep in mind that the integration
for lambda ranges from 0 to 1 for the forward direction and from 1 to 0
when integrating backwards)
gmx analyze -f dhdl.xvg -integrate
The integrated values, i.e. free energy estimates, may seem unexpectedly large,
however, note that "gmx analyze" integrated the values using simulation time to define integration ranges.
This means that dH/dl curve generated over 100 ps simulation is integrated from 0 to 100.
To get the correct integral in this case simply divide the obtained value by 100.
Go back to Contents
D1. Non-equilibrium calculations: simulations
In practice, however, the slow-growth TI is not frequently used.
An alternative method, that does not require an assumption of quasi-equilibrium during the alchemical transition
is the fast growth TI.
The fast growth TI approach relies on Jarzynski's equality (when transition is performed in one direction only)
or on the Crooks Fluctuation Theorem (when the transitions are performed in both directions).
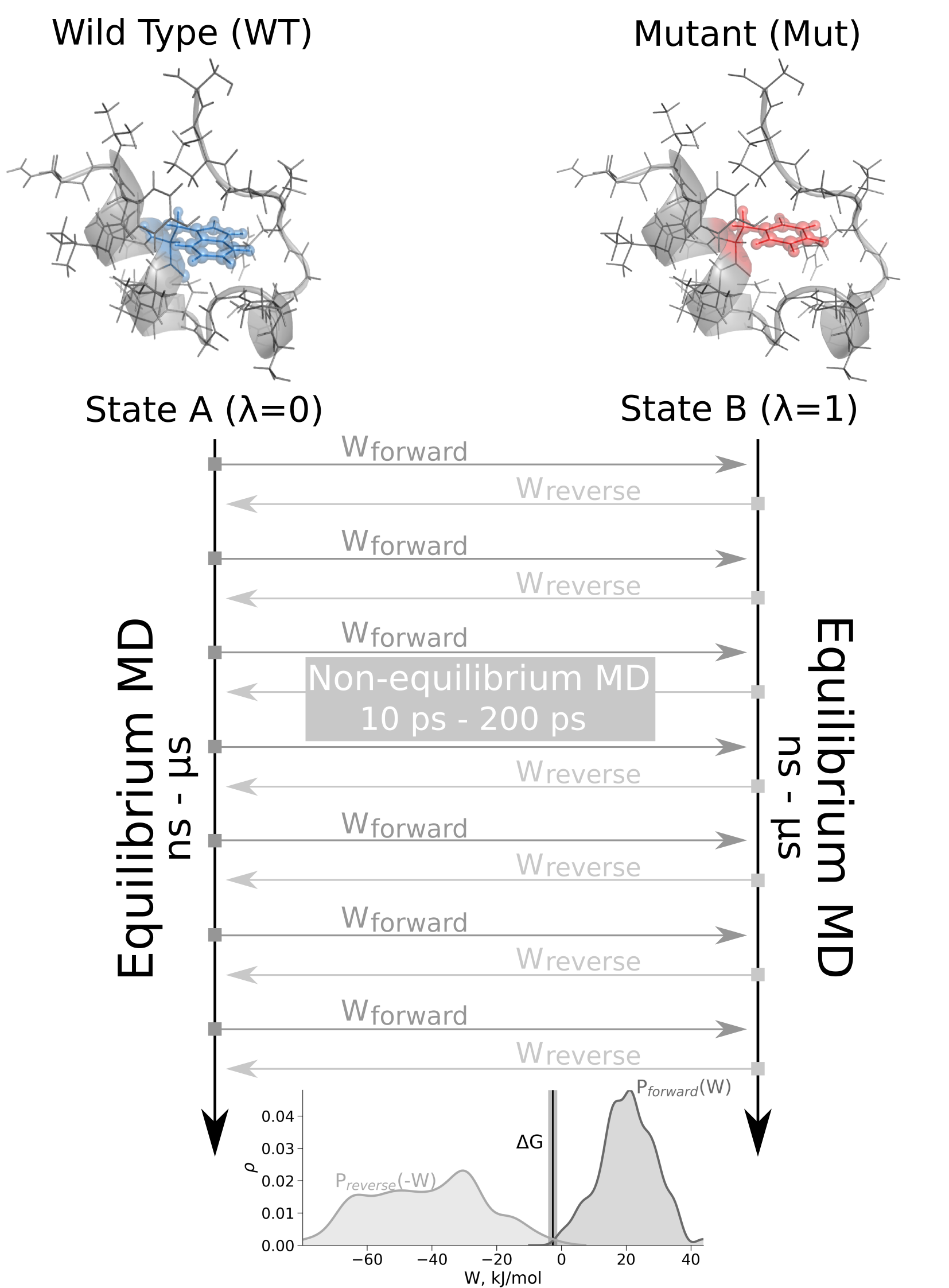
The schematic representation of the non-equilibrium alchemical free energy calculations is depicted on the right.
Here, free energy difference in the folded state of a protein is calculated.
Firstly, two independent equilibrium simulations need to be performed by keeping the system in its physical states: WT and mutant.
To start these simulations we need energy minimized structures in both states.
The energy minimized system in the WT state has already been obtained in one of the earlier steps and is available in the em.pdb file.
For the energy minimization in the mutant state use the emB.mdp file,
where init_lambda parameter sets the alchemical state to 1, i.e. mutant.
gmx grompp -f emB.mdp -c ions.pdb -p newtop.top -maxwarn 1
gmx mdrun -v -c emB.pdb
Next, prepare and run the equilibrium simulations in each of the states using eqA.mdp and
eqB.mdp.
These simulations need to sufficiently sample the end state ensembles, as the free energy accuracy will depend on the sampling convergence.
Typically the equilibrium simulations are in the nanosecond to microsecond time range.
However, to save time in the current tutorial we are using far shorter 100 ps simulations:
gmx grompp -f eqA.mdp -c em.pdb -p newtop.top -maxwarn 1 -o eqA.tpr
gmx grompp -f eqB.mdp -c emB.pdb -p newtop.top -maxwarn 1 -o eqB.tpr
gmx mdrun -v -s eqA.tpr -x eqA.xtc -ntomp 4 -ntmpi 1
gmx mdrun -v -s eqB.tpr -x eqB.xtc -ntomp 4 -ntmpi 1
From the generated trajectories snapshots are selected to start fast (typically 10 - 200 ps) transitions driving the system
in the forward (WT to mutant) and reverse (mutant to WT) directions.
Usually, to obtain accurate free energy estimates we use at least a hundred transitions in each direction.
To extract the snapshots gmx trjconv tool can be used as follows (here we are extracting only 5 snapshots in each direction):
echo 0 | gmx trjconv -s eqA.tpr -f eqA.xtc -o frameA.gro -pbc mol -ur compact -b 1 -sep -skip 20
echo 0 | gmx trjconv -s eqB.tpr -f eqB.xtc -o frameB.gro -pbc mol -ur compact -b 1 -sep -skip 20
Finally, for every for every generated snapshot we create a .tpr file and perform a transition simulation.
Here, for the purpose of illustration we use very fast 10 ps transitions for state A transitionA.mdp
and state B transitionB.mdp.
Below we only demonstrate the procedure for one frame.
To perform the same simulations for a large number (on the order of hundreds) of transitions
it is convenient to loop over the following commands in a script.
gmx grompp -f transitionA.mdp -c frameA0.gro -p newtop.top -maxwarn 1 -o transitionA0.tpr
gmx grompp -f transitionB.mdp -c frameB0.gro -p newtop.top -maxwarn 1 -o transitionB0.tpr
gmx mdrun -v -s transitionA0.tpr -dhdl dhdlA0.xvg -ntomp 4 -ntmpi 1
gmx mdrun -v -s transitionB0.tpr -dhdl dhdlB0.xvg -ntomp 4 -ntmpi 1
The work values required to perform these transitions are collected in the dhdl.xvg files and the Crooks Fluctuation Theorem is
used to calculate the free energy difference between the two states.
Go back to Contents
D2. Non-equilibrium calculations: analysis
The simulations performed above generated only several dH/dl curves for a few transitions.
To be able to estimate free energy difference we have pre-calculated more transitions.
The archive file S13A_fast_growth.tar.gz
contains two 10 ns simulation trajectories for the system in state A (eqA directory) and B (eqB directory).
(In case this file appears to be too large, you can download the file with analysis
folder only.)
From the trajectories 100 snapshots have been extracted and a fast 50 ps simulation has been performed
starting from each frame.
pmx has utilities allowing to integrate over the multiple curves and subsequently estimate free energy differences
using several approaches: Crooks Gaussian Intersection, Bennet Acceptance Ratio or Jarzynski's estimator.
Navigate to the extracted S13A_fast_growth directory and run
analyze_dhdl.py -fA eqA/morphes/frame*/dgdl.xvg -fB eqB/morphes/frame*/dgdl.xvg -t 300 --nbins 25
The free energy estimates are summarized in the file results.dat.
You can also investigate the individual work values for every transition in the forward (integ0.dat) and
backward (integ1.dat) directions. How do the non-equilibrium work values compare
to the quasi-equilibrium slow growth work estimates?
The script also produces a plot facilitating analysis of the work distributions.
So far we have investigated only one part of the cycle involving the folded protein.
To construct a double free energy difference the ΔG value for a mutation in the unfolded protein
(which we approximate by a GXG tripeptide) is needed.
The results for the A2S mutation in the tripeptide can be found here.
The final ΔΔG value will indicate whether the mutation is stabilizing the protein in its folded state.
Experimental measurements suggest that S13A mutation is mildly stabilizing (1.8 kJ/mol)
Barua et al, 2008.
Go back to Contents
Further references:
Books:
- Editors: Chipot C. , Christophe A. et al., Free Energy Calculations , Springer Series in Chemical Physics, Vol. 86, [link] .
- Vytautas Gapsys, Servaas Michielssens, Jan Henning Peters, Bert L. de Groot, Hadas Leonov. Calculation of Binding Free Energies. Molecular Modeling of proteins: 2nd edition. Book Series: Methods in Molecular Biology 1215: 173-209 (2015) [link]
Advanced reading:
- R. W. Zwanzig, High Temperature Equation of State by a Perturbation Method. I. Nonpolar Gases J. Chem. Phys. 22: 1420 (1954). [link]
- Goette M, Grubmuller H, Accuracy and convergence of free energy differences calculated from nonequilibrium switching processes. J. Comp. Chem. 30: 447-456 (2009). [link]
- Gapsys V, Michielssens S, Seeliger D, and de Groot BL pmx: Automated protein structure and topology generation for alchemical perturbations. J. Comp. Chem. 36:348-354 (2015) [link]
- Vytautas Gapsys, Servaas Michielssens, Daniel Seeliger and Bert L. de Groot. Accurate and rigorous prediction of the changes in protein free energies in a large-scale mutation scan. Angew. Chem. Int. Ed. 55: 7364-7368(2016) [link]
- Seeliger D, and de Groot BL Protein thermostability calculations using alchemical free energy simulations. Biophys. J. 98:2309-2316 (2010) [link]
Go back to Contents