- General description
- Structure file
- Structure check
- Force field selection
- Number of mutations
- Amino acid number
- Terminal residues
- Scan
- Amino acid code
- Hydrogens
- Output
- Procedure and error report
- Terms of use
General description
This webserver is a front-end for a pmx based hybrid protein structure and topology generation
for alchemical molecular dynamics based free energy calculations.
The created structures and topologies are compatible with the Gromacs molecular dynamics
simulation software.
To top
Structure file
Only .pdb files are supported.
The webserver will only retain the protein moiety, i.e. ligands or other types of molecules will be discarded.
Residue naming will be adjusted according to the selected force field.
If there are missing atoms they will not be added and the submission will result in an error.
To reconstruct the missing parts of the structure you may try
Rosetta, Modeller or WHAT IF.
It is also recommended to perform a structure checking step: Check (opt.) button.
This option allows testing whether the selected structure would be properly processed by the webserver.
To top
Structure check
This is an optional step - "Check(opt.)" button - to check whether the structure file would be successfully processed by the webserver.
It quickly runs the structure pre-processing steps without requiring to submit the whole query.
In case of an error, information about the source of the problem is provided.
A successful structure check also provides some additional information which may be important when submitting the final query.
For example, if pdb2gmx cannot understand hydrogen names in the system, the box "Use pdb2gmx to assign hydrogens?" would need
to be set: the structure checking option would inform about this situation (see Webserver tutorial).
To top
Force field selection
Currently 5 force fields are available.
Upon request, support for more force fields may be possible.
To prepare simulations with the hybrid topologies
download these force fields and set GMXLIB variable to the "mutff45"
directory:
mutff45.tar.gz
To top
Number of mutations
An arbitrary number of simultaneous mutations in a protein are allowed.
Interactively up to 3 mutations can be selected.
For more than 3 mutations a web-based form needs to be filled.
In the following example 4 residues will be mutated.
Note that the residues do not need to be entered sequentially.
The input may contain two or three values per entry.
If three values are entered, the first character will be interpreted as a chain identifier and the second value will mark residue ID.
For the entries with two values, the first value will be interpreted as a residue ID and the first residue in the structure matching this ID will be used.
The last value in every entry denotes the mutation to be performed.
Example:
X 3 A
Y 5 V
20 R
A 11 D
To top
Amino acid number
An amino acid to be mutated can be defined by a chain ID together with a residue number.
Chain ID is optional: if not given, the webserver will search for the first residue in the structure that has a number matching the provided one.
To top
Terminal residues
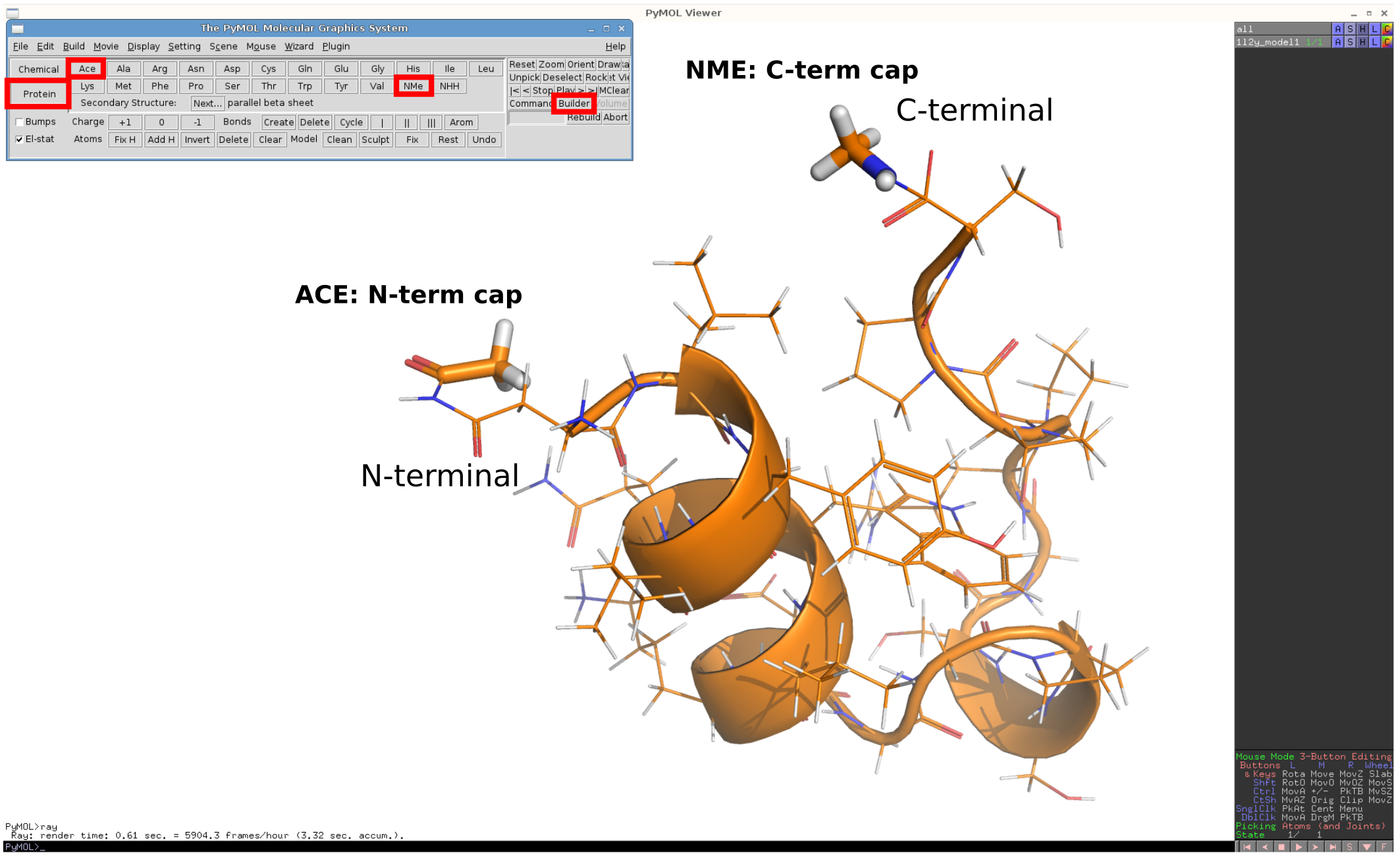
Mutations of the terminal residues are not supported.
If a terminal residue is to be mutated, adding a capping group to the terminal residue would technically make the amino acid not terminal any more,
and the mutation can be performed.
Pymol allows for a convenient addition of capping residues:
after loading a structure select "Builder" and in an opened menu select "Protein".
Select a capping group ACE for the N-terminal residue.
By combining this selection with the existing object simply click on the nitrogen atom of the N-term amino acid.
For the C-terminal cap, use NME residue and combine it with the carboxyl carbon of the C-terminal amino acid.
You can see an illustration of the procedure in
this image.
To top
Scan
If a scan option is selected, the webserver will mutate every residue in the protein by the user selected amino acid.
If a chain ID is provided, only the residues in the specified chain will be mutated.
As a result of a scan, a separate folder for every mutation will be generated containing the respective hybrid structure and topology.
The webserver will discard unsupported or irrelevant mutations, e.g. proline mutations, glycine mutations in the Charmm family force fields
or mutations to the same amino acid, disulfide bridge forming cysteine mutations, terminal residues.
To top
Amino acid code
Mutation has to be indicated by a single letter code:
| Amino acid | Code |
| Alanine | A |
| Arginine | R |
| Asparagine | N |
| Aspartate | D |
| Aspartate protonated | B |
| Cysteine | C |
| Glycine | G |
| Glutamate | E |
| Glutamate protonated | J |
| Glutamine | Q |
| Histidine: only Nε2 protonated | X |
| Histidine: only Nδ1 protonated | H |
| Histidine: Nε2 and Nδ1 protonated | Z |
| Isoleucine | I |
| Leucine | L |
| Lysine | K |
| Lysine unprotonated | O |
| Methionine | M |
| Phenylalanine | F |
| Serine | S |
| Threonine | T |
| Tryptophan | W |
| Tyrosine | Y |
| Valine | V |
Proline mutations are not supported.
Glycine involving mutations in the Charmm force fields are not supported because alchemical CMAP transitions are not yet available in Gromacs.
Disulfide bridge forming cysteine mutations are not supported to avoid alchemical bond breaking.
To top
Hydrogens
By default hydrogens are generated using pdb2gmx functionality.
If this option is de-selected hydrogens as provided in the structure
file will be retained.
Note, however, that in the latter case atom names in the structure
must match those in the selected force field.
To top
Output
Hybrid structure is provided in the hybrid.pdb file.
Hybrid topology for a single chain protein is in the hybrid.itp file.
For a multi-chain protein every chain will have a topology .itp file
generated: hybrid_Protein_chain_X.itp (here X denotes chain identifier).
The .itp topologies are collected in a single hybrid.top file.
The output is provided as a single file in a compressed format .tar.gz
which will be kept on the server for 7 days.
To top
Procedure and error report
At first a number of scripts will check and pre-process the input parameters,
structure file and, if provided, mutation list file.
Afterwards, the actual hybrid structure and topology generation consisting of 4 steps
starts.
Step 1: pdb2gmx is run to obtain atom and residue naming in a structure file compatible with the
user selected force field. If hydrogen generation was selected, it will be performed in this step.
Step 2: mutate.py will use the structure from the previous step
to generate a new hybrid structure with the selected mutations.
Step 3: pdb2gmx is run again. This time the topology for a hybrid structure is generated.
Step 4: generate_hybrid_topology.py adds a B-state to the hybrid topology.
If an error occurs at a pre-processing step, a brief message will be printed.
An error in any of the 4 steps of the main procedure will indicate the step number
and the full error message generated.
To top
Terms of use
The mutation libraries and generated hybrid structures/topologies
can be used without any restrictions.
Please note that the authors provide no warranty of any kind for using
these files.
To top